
山西煤化所在金屬間電子化物表面自發水解離促進CO?甲烷化研究中取得重要進展
近日,中國科學院山西煤炭化學研究所溫曉東團隊在理論-實驗-數據一體化研究思路指導下,針對金屬間電子化合物(IE)催化CO?甲烷化研究取得重要進展,研究成果以“Spontaneous water dissociation on intermetallic electride LaCu0.67Si1.33?enhances electrochemical methanization of CO2”為題發表在《Nature Communications》期刊上。
利用可再生電力驅動的二氧化碳(CO2)電還原轉化為甲烷是一種可持續的減少對天然氣依賴的方法。然而,這一過程目前受限于效率和耐久性不足,這主要源于現有催化劑上水解離(WD)和質子耦合電子轉移之間的動力學差異。為了提高CO2電還原為甲烷的效率,研究者們致力于設計更高效的催化劑,特別是銅(Cu)基催化劑。盡管如此,如何通過催化劑設計來同步實現高效的水解離和CO2活化仍然是一個挑戰。
團隊基于自研的機器學習加速結構預測方法(J. Phys. Chem. C, 2020,124,17244?17254),結合基于密度泛函理論(DFT)的高通量計算,揭示了金屬間電子化合物材料關鍵活性位的富電子結構,并進一步預測其具有優異的自發水解離能力。結合LaCu0.67Si1.33材料制備并設計表征實驗,利用H2O-程序升溫表面反應(H2O-TPSR)證實了在該金屬間電子化合物表面發生了顯著的自發水解離現象。在堿性流通池中,IE LaCu0.67Si1.33催化劑在相對于可逆氫電極(vs. RHE)?1.21 V的還原電位下表現出72%的甲烷法拉第效率(FE),并在?1.52 V vs. RHE下達到476.7 mA cm?2的甲烷偏電流密度。密度泛函理論計算進一步表明,在IE LaCu0.67Si1.33催化劑上,甲烷化反應路徑在熱力學/動力學上更為有利。
通過數據挖掘,結構表征及理論預測,團隊揭示了IE LaCu0.67Si1.33催化劑獨特的活性位富電子結構和自發水解離能力,為CO2的電化學甲烷化以及其他可能涉及水解離過程的可再生能源轉化,提供了一個極具前景的催化劑體系。以上工作得到國家科技部重點研發計劃(批準號:2022YFA1604100),國家自然科學基金杰出青年基金(批準號22225206),中國科學院前沿科學重點研究項目(ZDBS - LY - 7007)等項目的支持。
本成果由中國科學院山西煤炭化學研究所溫曉東團隊和北京大學駱明川團隊合作完成。
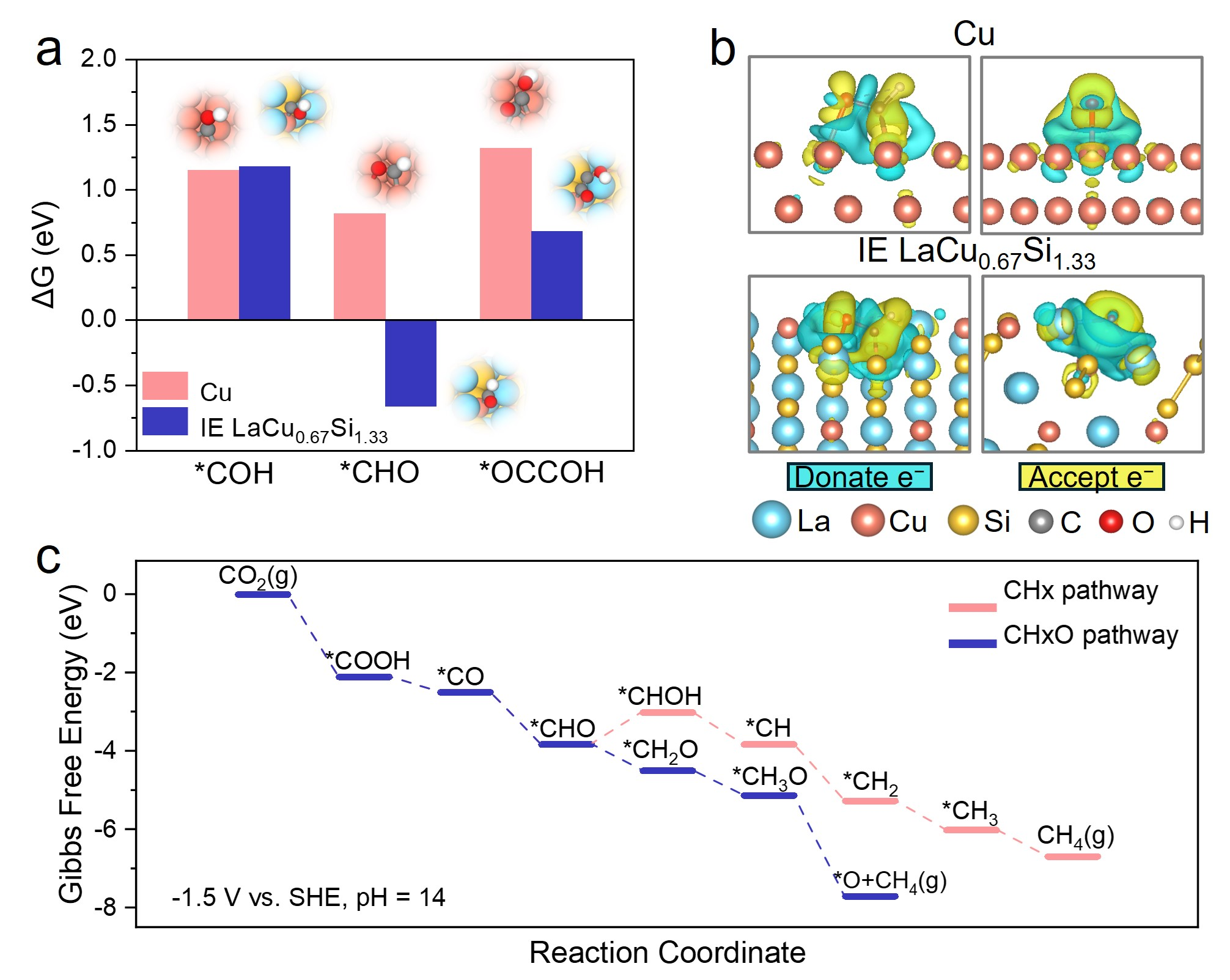
DFT計算(a) *CO反應系列中間物種在IE LaCu0.67Si1.33和Cu表面加氫和碳碳耦合反應的吉布斯自由能;(b)*CHO反應中間物種在IE LaCu0.67Si1.33和Cu表面的電荷密度差圖;(c)CO2-CH4轉變不同反應路徑中間過程的吉布斯自由能。
(煤炭高效低碳利用全國重點實驗室)
附件下載:



